MANEJO DEL NEUMOTORAX
AUTORES:
Maria del Mar Vazquez Jiménez
Carlos Rueda Rios. Especialista en Neumología
Correspondencia con:
Carlos Rueda Rios
Hospital Universitario Virgen de la Victoria
MALAGA
Campus de Teatinos s/n
INDICE
I. Definición.
II. Clasificación.
III. Neumotórax espontáneo primario
1. Epidemiología
2. Clínica
3. Exploración física
4. Pruebas complementarias
5. Complicaciones
6. Tratamiento.
IV. Apéndice 1. Colocación del drenaje pleural.
V. Apéndice 2. Técnica de inserción del drenaje pleural.
VI. Apéndice 3. Manejo y criterios de retirada del drenaje pleural.
VII. Apéndice 4. Complicaciones del drenaje pleural.
VIII. Figuras.
IX. Bibliografía
X. Algoritmo
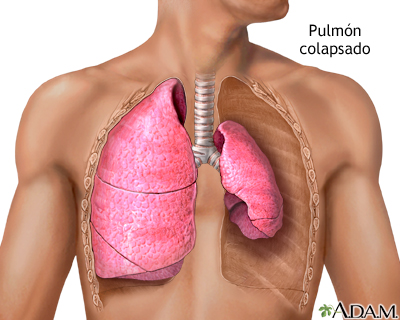
I. DEFINICION
El neumotórax se define como la presencia de aire dentro de la cavidad pleural que provoca el
colapso pulmonar del tejido adyacente.
II. CLASIFICACIÓN
Los podemos clasificar en función de la causa que los ha producido y en función de la
enfermedad pulmonar de base. Pueden ser espontáneos, traumáticos o yatrogénicos en
función de la causa y a su vez los espontáneos los podemos dividir en primarios o secundarios
en función de la patología respiratoria (tabla 1).
II.1 El neumotórax traumático se divide en cerrado o abierto en función de la
presencia o no de herida penetrante, con mucha frecuencia se acompaña de hemotórax de
cuantía variable. El barotrauma suele darse en pacientes sometidos a ventilación mecánica,
relacionándose este hecho con el uso de volúmenes corrientes y PEEP elevados.
II.2 Si no nos encontramos ante una causa clara estamos ante un neumotórax
espontáneo.
Yatrogénico:
• Tras cateterización de vías centrales, biopsia pleural,
toracocentesis, PAAF
Traumático:
• Heridas abiertas o cerradas
• Barotrauma
Espontáneo:
• Primario o idiopático
• Secundario (lesión pulmonar previo)
A/ Hablamos de neumotórax espontáneo primario si no encontramos ninguna
causa pulmonar de base que lo justifique y lo desarrollaremos más adelante.
B/ neumotórax espontáneo secundario, encontramos diversas situaciones
patológicas con afectación pulmonar, como son:
a) Asma: suele coincidir con las agudizaciones y con frecuencia se acompaña
de neumomediastino.
b) EPOC: con frecuencia son secundarios a la rotura de bullas intrapulmonares.
Suele cursar con importante afectación general y es conveniente sospecharla en
todo EPOC con aumento brusco de su disnea crónica y dolor pleurítico.
c) Fibrosis pulmonares avanzadas: en aquellas que existe panalizacion y bullas
como en el granuloma eosinófilo.
d) Enfermedades del tejido conectivo como síndrome de Marfan y Ehlers-
Danlos.
e) Neumotórax catamenial es una entidad rara que se da en mujeres de mediana
edad en el contexto de la menstruación y su sustrato anatomopatológico
corresponde a un foco endometriósico ectópico.
f) En cuanto a las causas infecciosas hay que destacar las neumonías
necrotizantes en especial la originada por el estafilococo, la tuberculosis que
puede originar focos caseosos subpleurales, el pneumocystis carinii , ...
g) Otras asociaciones son lo la fibrosis quiísticas, neumonías aspirativas,
infecciones fúngicas, sarcoidosis, carcinoma broncogenico, metástasis, etc.
III. NEUMOTORAX ESPONTANEO PRIMARIO
La causa más frecuente de rotura de pequeños blebs, que son colecciones de aire
subpleural resultado de ruptura alveolar, el aire diseca los tejidos conectivos adyacentes, acumulándose entre la lámina interna y externa de la pleura visceral. Los blebs se hallan con
más frecuencia en el vértice del lóbulo superior o en el vértice posterior del lóbulo inferior.
El neumotórax espontáneo es independiente del esfuerzo o la tos y se suele ver en varones
entre 20 y 40 años con tipo asténico y con hábito tabáquico. Se estima una incidencia de 7,4-
28 casos/100.000 habitantes en hombres y de 1.2-10 casos/100.000 habitantes en mujeres.
Es más frecuente en el lado derecho, los neumotórax bilaterales se producen en menos del
10% de los pacientes, en los dos primeros años recurren un 25% de los pacientes. Después del
segundo neumotórax, la posibilidad de tener un tercer episodio aumenta a más del 50%.
Las complicaciones más frecuentes en el desarrollo del neumotórax son el derrame pleural no
complicado en el 20%, el hemotórax con cuantía significativa en menos del 5% , el
neumotórax a tensión en un 2-3%, a mucha distancia el enfisema subcutáneo, el
neumomediastino , el empiema o la cronificación.
Las complicaciones postratamiento más frecuentes son la recurrencia y la fuga aérea
persistente que son indicaciones de cirugía.
III.1. CLINICA
Los síntomas depende de dos factores la reserva respiratoria del paciente y el tamaño
de neumotórax. Debido a que muchos de ellos son de pequeño tamaño y suceden en pacientes
sin patología pulmonar previa, pueden no provocar disnea debido a la reserva funcional del
paciente. Se estima que en un 5-10% de los casos pueden ser asintomáticos.
El síntoma más frecuente es la región torácica lateral, dolor en punta de costado, se
puede irradiar al resto del tórax y al cuello. Suele ser intenso, de carácter punzante y de inicio
agudo, con duración variable. Suele aumentar con la tos y movimientos respiratorios
profundos y la tos, aliviándose con la respiración superficial e inmovilización.
En algunos casos pueden existir manifestaciones vegetativas (sudoración, taquicardia,
palidez) u otro tipo de síntomas con tos seca, hemoptisis, síncope y debilidad de miembros
superiores.
III.2. EXPLORACIÓN FISICA
Cuando el tamaño del neumotórax es significativo encontramos disminución de los
movimientos de la pared del lado afecto, timpanismo a la percusión y disminución e incluso
ausencia de los ruidos respiratorios a la auscultación. En ocasiones podemos auscultar roce
pleural y taquicardia.
III.3. PRUEBAS COMPLEMENTARIAS
El diagnóstico clínico se confirma por radiografía posteroanterior y lateral de tórax al
identificar la línea del margen de la pleura visceral, separada de la pleura parietal existiendo
entre ambas un espacio aéreo hiperclaro sin trama vascular, de manera que el pulmón
adyacente se observa más denso a permanecer parcialmente colapsado (figura 5). En
neumotórax pequeños se puede realizar una posteroanterior de tórax en espiración forzada,
que identificará más fácilmente la línea de la pleura visceral. Es frecuente observar un
pequeño nivel hidroaéreo en el seno costofrénico, que no tiene ninguna relevancia clínica.
En ocasiones podemos observar que parte del parénquima permanece unido a pleura
parietal gracias a adherencias pleurales (figura 2 y 3)
En el electrocardiograma podemos observar una serie de signos característicamente
reversibles. En pacientes con neumotórax izquierdos el EKG puede mostrar una desviación a
la derecha del eje, con disminución de la amplitud del QRS e inversión de la onda T en
derivaciones izquierdas. En grandes neumotórax con la interposición de gas entre el corazón y
el electrodo puede producir cambios electrocardiográficos que simulen infartos anteriores.
III.4. COMPLICACIONES
A/ Neumotórax a tensión:
Se produce a raíz de que el aire pase del pulmón al espacio pleural durante la
inspiración y no salga por un mecanismo valvular. A medida que la presión en
el hemitórax aumenta, el mediastino se desplaza al lado contralateral interfiriendo con la ventilación, dificultando el retorno venoso y en último caso
disminuyendo el gasto cardiaco.
Debemos sospecharlo, cuando existe disnea intensa progresiva, taquipnea,
cianosis, taquicardia, hipotensión, diaforesis y distensión de las venosa
cervical.
Radiológicamente se observa desplazamiento contralateral de la tráquea y del
mediastino y depresión del diafragma ipsilateral
Si se sospecha el diagnóstico es imperativo el drenaje con aguja, catéter o tubo
sin confirmación radiográfica, antes de que se produzca el colapso circulatorio
completo. Una vez que ha sido solucionada la urgencia por la tensión, el
tratamiento ulterior debe ser similar al resto de pacientes con episodio
complicado.
B/. Hemoneumotórax:
Es una complicación poco frecuente, que se produce por rotura de
adherencias entre pleura parietal y visceral vascularizadas al producirse el
colapso pulmonar. A menudo la reexpansión pulmonar con drenaje ayuda a
taponar el lugar de sangrado. Si el sangrado no se controla o no se reexpande el
pulmon por la presencia de coágulos hay que valorar cirugía.
C/. Neumomediastino:
Se debe a que el aire pasa al mediastino que diseca a lo largo de los
bronquios y vainas vasculares de los vasos pulmonares, generalmente no tiene
consecuencias clínicas. No obstante debe descartarse otras causas como rotura
de la vía aérea o perforación de esófago.
D/.Enfisema subcutáneo:
No suele tener una implicación clínica significativa, si se produce tras el
drenaje indica que éste está mal posicionado u obstruido, o bien uno de los
orificios de drenaje se halla en pared torácica.
E/.Neumotórax bilateral:
El neumotórax bilateral simultáneo es raro (menor del 1%) y precisa, en
la fase aguda, drenaje de ambos espacios pleurales. Es más frecuente el
neumotórax bilateral secuencial (recidiva contralateral) que es indicación.
F/.Pioneumotórax
Generalmente es secundario a neumonía necrotizante o a una rotura
esofágica. Es indicación de drenaje urgente.
G/.Neumotórax crónico
Se trata de un neumotórax que persiste más de tres meses. Suelen
perdurar gracias a la existencia de adherencias pleurales, fístula broncopleural
a través de una bulla o alteración parenquimatosa (necrosis, nódulo, etc). La
cavidad se suele rellenar de líquido. Suele tratarse con intervención quirúrgica.
III.5. TRATAMIENTO
Va a depender de varios factores: tamaño del neumotórax, enfermedad pulmonar
previa, causa, síntomas recidiva, tratamientos previos, profesiones de riesgo, etc., puede ir
desde la observación hasta el abordaje quirúrgico. Las recidivas ocurren en un 30 a 50% de
los NEP, y en un 80% de los casos se producen durante el primer año.
El tratamiento debe cumplir dos objetivos:
1-evacuar el aire de la cavidad pleural
2-toda vez que el aire halla sido drenado, conseguir una reexpansión duradera y
estable que evite las recidivas.
Hay una serie de medidas generales que hay que imponer a todo neumotórax sea cual
sea su tamaño y es el reposo y la oxigenoterapia, que ayudan a acelerar la reabsorción del aire
intrapleural.
En neumotórax espontáneo primario podemos ofrecer cinco tipos distintos de tratamiento:
• Reposo y observación clínica. en régimen de ingreso hospitalario o en domicilio
paciente colaborador, proximidad geográfica, cuando se trata de un primer episodio de
neumotórax espontáneo primario, menor del 20% y asintomático.
• Drenaje pleural: indicado en el primer episodio de los NEP mayores del 20% y en
todos los neumotórax espontáneos secundarios.
• Pleurodesis
• Cirugía mediante toracotomía con pleurectomía parietal parcial o abrasión mecánica
• Cirugía videotoracoscópica.
1. Reposo y observación clínica.
Cuando se trata de un primer episodio de neumotórax espontáneo primario, menor del
20% y asintomático, en pacientes sin enfermedad respiratorio de base pueden ser tratados de
forma expectante con reposos y preferiblemente observación hospitalaria durante las primeras
24-48 horas. Posteriormente se hará un seguimiento clínico-radiológico ambulatorio.
Hay grupos que están planteando en tratamiento expectante en domicilio. Se lleva a cabo
en paciente colaborador y con criterios de proximidad geográfica, deberá permanecer en
reposo con oxigenoterapia continua con posibilidad de seguimiento en consulta.
El principal inconveniente de esta opción terapéutica es la progresión del neumotórax o la
no reexpansión, en cualquier caso si pasado una semana el neumotórax ha progresado o no se
ha reexpandido el pulmón se recomienda colocación de drenaje.
2. Drenaje pleural:
Indicado en el primer episodio de los NEP mayores del 20% , en los a tensión en herida
abierta del tórax, bilateral, sintomático, enfisema subcutáneo, neumomediastino, progresión radiográfica, caso de ventilación mecánica y en todos los neumotórax espontáneos
secundarios.
La técnica de inserción, seguimiento y criterios de retirada se explicaran más adelante
Los tubos de drenajes finos, pueden utilizarse en neumotórax espontáneo primario, sobre
todo si es el primer episodio y si el neumotórax no se acompaña de traumatismo, hemotórax,
hidrotórax ni disfunción respiratoria. Los tubos de drenajes se conectan a sistemas
unidireccionales que no permitan la entrada de aire en cavidad pleural, el más utilizado es el
de tres cámaras.
3. Pleurodesis:
Se reserva para pacientes que no puedan ser sometidos a toracotomía (mala calidad de
vida previa, edad avanzada, neoplasias con progresión pleural,...). Su objetivo es conseguir a
unión de ambas pleuras con distintos agentes (talco, clorhidrato de tetracicilina, bleomicina o
colas biologicas) que se administran mediante drenaje pleural o toracoscopia.
4. Cirugía:
Se realiza por minitoracotomía axilar con pleurectomía parietal parcial o abrasión pleural,
que a diferencia de la anterior preserva el plano extrapleural. En los últimos años gracias al
desarrollo de las suturas mecánicas la resección de bullas es mucho más efectivo.
5. Cirugía videotoracoscópica (CVT):
Con resultados similares a la cirugía convencional, pero es menos agresiva y tiene un
periodo postquirúrgico más corto. Consiste en la realización de por lo menos tres orificios en
el hemitórax afecto por los que se introduce una cámara y el instrumental.
Las ventajas de la cirugía videotoracoscopica se puede cifrar en cuatro:
1. Permite una mejor visualización de toda la superficie pleural
2. Es más rápida de realizar.
3. Produce menos dolor postoperatorio
4. Disminuye la estancia hospitalaria.
APENDICE 1. COLOCACIÓN DE DRENAJE PLEURAL
SECUENCIA MATERIAL
PREPARACIÓN
Explicación
Posición sentado o decúbito supino
Atropina 0,5 mg IM
Localización del sitio de punción
Marcar el lugar de punción
CAMPO ESTERIL
Povidona yodada, paño, guantes y gasas
esteriles
ANESTESIA LOCAL
Agujas IM, Jeringas de 10 ml,
Escandicaina
MATERIAL DE DRENAJE
Tubo de drenaje: convencionales de silastic con múltiples orificios, con línea radiopaca hasta el último agujero de varios diámetros, tipo Pleurecath catéter fino y largo con llave de tres pasos y el Neumovent catéter fino y corto que lleva incorporada una válvula unidireccional, pinzas de Kelly, sutura, porta, sistema de sellado de agua y aspirador.
APENDICE 2. Técnica de inserción del tubo de drenaje.
Se coloca al paciente sentado o en decúbito supino se elige la línea medioclavicular en
2ª espacio intercostal (aire) o en línea axilar media 5ª-6ª espacio intercostal (liquido o aire).Se
realiza la limpieza y antisepsia de la piel.
Los tubos finos tienen un fiador interno o externo y sólo tienen que ser empujados a
través de la zona previamente anestesiada hasta llegar a cámara pleural. Se dirige la punta del
catéter al apice y se extrae el fiador. Se fija en tubo con un punto y se conecta al sistema de
vacío a través de la llave de tres pasos.
En el tubo de drenaje convencional se anestesia con rutina habitual y se realiza una
incisión en piel y se diseca por planos con pinza de Kelly debiendo ir siempre por borde
superior de la costilla. Se introduce suavemente el tubo de drenaje, se debe comprobar
presiones intrapleurales, fijar drenaje, se conecta a sistema de sellado de agua (pleurevac) y se
comprueba colocación mediante radiografía.
Para el líquido se prefiere una localización en 5º o 6º espacio intercostal en línea axilar
media, deben evitarse localizaciones más bajas por el riesgo de atravesar diafragma y lesionar
órganos intraabdominales, evitaremos si es posible la colocación en la espalda, ya que
técnicamente es más complicado y produce un innecesario dolor al enfermo.
Posteriormente se conecta a aspirador caso de tratarse de un neumotórax o si no evacua de
forma efectiva en derrame pleural (nunca mas de 1000 ml/8h)
APENDICE 3. Manejo y criterios de retirada de drenaje pleural
Manejo
Tras la inserción del tubo se comprueba posición y la marcha de la reexpansión pulmonar con
una radiografía. Debe ser conectado a un sistema que impida la salida de aire pero no su
entrada, estos sistemas se dividen en dos bloques: pasivo (válvula de Heimlich) o activo
(recolector de tres cámaras) El tubo nunca debe ser pinzado para el traslado de los enfermos,
especialmente si tiene fuga aérea por el peligro de que se convierta en neumotórax
hipertensivo. .
Si un drenaje torácico se desconecta accidentalmente simplemente debe ser reconectado.
Las pequeñas oscilaciones de los líquidos del tubo indican que están permeables si no se
mueven o el pulmón se halla reexpandido o el tubo obstruido. Grandes oscilaciones se asocian
a atelectasia o reexpansión incompleta.
Para repermeabilizar un tubo puede ordeñarse, pero no se recomienda la irrigación con suero
salino por el peligro de contaminación.
Criterios de retirada
Utilizamos tres criterios clásicos fundamentales :
• no hay oscilaciones de líquido
• el drenaje es menor de 100 cc por día
• no hay fuga aérea.
Se pinza el tubo durante 24 horas y si no se ha reproducido el neumotórax se extrae el
tubo. Para cerrar el orificio debe anudarse el punto que se dejó con este propósito (tubos de
calibre convencional) o gasas con vaselina en los tubos de menor calibre. Tras la retirada debe
hacerse una radiografía de control.
Sistema de tres cámaras:
La primera recoge el líquido, la segunda actúa como sello de agua que no permite la
entrada de aire y permite su salida y la tercera es la cámara de control de succión, que en los
sistemas secos se ha sustituido la columna de agua por un sistema valvular.
Este sistema se conecta a su vez a la red de presión negativa del hospital o a los sistemas
portátiles de aspiración.
APENDICE 4. Complicaciones de los drenajes
a) Fallo de la reexpansión y cronicidad: Nos ocurrirá caso de reexpansión incompleta a pesar
de la buena colocación y aspiración del tubo. En este caso se requiere toracotomía para
valorar la decorticación.
b) Fuga aérea persistente: Ocurre en aproximadamente un 4% de los drenajes. Generalmente
si esta situación se prolonga más de 7-10 días habría que valorar cirugía, caso de que se
acompañe de reeexpansión incompleta hay que valorar un segundo drenaje.
c) Colocación incorrecta: Lo mas frecuente es en los tubo de pequeño calibre al dejar algún
orificio de drenaje fuera de cavidad pleural, provocando enfisema subcutáneo, más
infrecuente es colocarlo en los tejidos blandos de cavidad torácica. En cualquiera de estas dos
situaciones habría que recolocar el tubo.
c.1 Caso de perforación pulmonar, diafragma u órgano intraabdominal habrá que
valorar cirugía urgente.
c.2 Caso de colocar un tubo dentro de una bulla habrá que operar al paciente
inmediatamente.
d) Enfisema quirúrgico: casi siempre ocurre por una colocación incorrecta del tubo o bien por
obstrucción del sistema de aspiración.
e) Hemorragia intrapleural: Suele ocurrir más en ancianos y se da por colocar en tubo justo
bajo la costilla.
f) Empiema: Se produce más en neumotórax secundarios a traumatismos o casos de drenajes
mantenidos durante tiempos prolongados.
g) Edemas de reexpansión: Es una complicación rara pero potencialmente letal. Se produce
cuando el aire o el líquido se evacuan demasiado rápido por un aumento de la permeabilidad
capilar. Los síntomas son tos progresiva, dolor pleurítico y edema bilateral. Para prevenirlo
debemos pinzar el tubo periódicamente para evitar la reexpansión súbita.
h) Neuralgia intercostal: Presenta dolor irradiado anteriormente, y en ocasiones se cronifica
mucho después de la retirada del drenaje.
FIGURA 1
BULLA VISUALIZADA POR TORACOSCOPIA.
16
FIGURA 2
Figura 2. ADHERENCIAS PLEURALES
17
FIGURA 3
NEUMOTORAX SECUNDARIO CON
ADHERENCIA PLEURAL.
18
FIGURA 4
NEUMOTÓRAX DEL 100% PULMON IZQUIERDO CON
DESPLAZAMIENTO MEDIASTINICO
CONTRALATERAL.
19
FIGURA 5
NEUMOTORAX ESPONTÁNEO PRIMARIO.
20
fIGFIUGRURA 6
DISPOSITIVO DE DRENAJE DE 3 CAMARAS.
21
FIGURA 7
Esquema de funcionamiento de la válvula unidireccional de Heimlich.
22
BIBLIOGRAFIA
• Baumann MH, Strange C. The Clinician's Perspective On Pneumothorax Management.
Chest. 1997; 112(3). 822-8.
• Ginferrer Garolera JM, Fernander-Retana P, Rami Porta R. Tratamiento del neumotorax
espontáneo mediante drenajes de pequeño calibre. Arch Bronconeumol. 1990; 26 144-
146
• Sanchez-Lloret J. Canto A, Borro Jm, Gimferrer JM. Diagnóstico y tratamiento del
neumotórax. Arch Bronconeumol 1998; 34 Supl 3:24-30
• Schramel FM, Postmus PE, Vandrschueren RG. Current aspects os spontaneous
pneumothorax. Eur Respir J 1997; 10: 1372-1379
• Sahn SA, Heffner Spontaneuous pneuomothorax. N Engl J Med 2000; 342:868-873
• Rueda Ríos C, Tibos F Hidalgo Sanjuán MV. Hemoptisis catamenial. A propósito de un
caso. Arch Bronconeumol. 2000; 36: 539
23
CUESTIONARIO
1. En un paciente con clínica de dolor de carácter pleurítico y disnea de aparición súbita, sin
antecedentes respiratorios de interés en un centro de salud que aporta radiografía de tórax
AP en inspiración normal. En la exploración la auscultación es normal. Cual sería la
actitud más correcta:
a) Oxigenoterapia y broncodilatadores.
b) Colocar un drenaje pleural tipo abocath en hemitórax afecto.
c) Tratar con benzodiacepinas
d) Realizar radiografía de tórax en espiración forzada
e) Realizar electrocardiograma.
2. Un enfermo con un cáncer de colon que precisa la canalización de una vena subclavia para
la su ciclo quimioterápico, comienza a presentar disnea y dolor torácico de carácter
pleurítico. En la exploración el enfermo presenta cianosis, frecuencia respiratoria de 35
rpm y disminución del murmullo vesicular en hemitórax derecho ¿diagnostico más
probable?
a) Reacción alérgica a la quimioterapia.
b) Tromboembolismo pulmonar
c) Angina
d) Neumotórax
e) Trombosis de la vena subclavia.
3-Paciente de 16 años que presenta un neumotórax izquierdo de menos de un 15% sin
repercusión respiratoria. Hace 2 años presentó un neumotórax derecho. Señale cual sería el
tratamiento más adecuado:
a) Pleurodesis química con talco
b) Drenaje torácico
c) Observación y cirugía posteriormente
d) Profilaxis antibiótica para evitar la infección de la cavidad pleural.
f) Pleurodesis con urokinasa.
24
4-Paciente de 20 años neumotórax con colapso pulmonar del 40%, con antecedente de un
episodio previo del mismo lado:
a) Drenaje torácico
b) Drenaje torácico y posterior intervención quirúrgica
c) Pleurodesis química con talco
d) Observación durante 24 horas y si el colapso pulmonar no aumenta al alta domiciliaria
e) Oxigenoterapia de alto flujo.
5-A los 20 minutos de una intervención quirúrgica en la que se requirió ventilación mecánica
y aporte de fluidos por vía central , un enfermo presenta tos seca, disnea y sensación de
opresión torácica súbitamente. En la exploración se aprecia disminución del murmullo
vesicular en hemitorax derecho. Diagnóstico.
a) Atelectasia masiva
b) TEP
c) Neumotórax
d) IAM
e) Crisis de ansiedad postquirúrgica
6- En relación al neumotórax, es FALSO que
a) Los neumotórax tienen una alta frecuencia de recidivas.
b) Los neumotórax espontáneos primarios suelen ocasionarse por rotura de blebs
apicales.
c) La causa más frecuente de neumotórax espontáneo secundario es EPOC.
d) Ante la sospecha de neumotórax debe solicitarse una radiografía en inspiración
forzada
e) Los neumotórax espontáneos primarios no tienen relación con el hábito tabáquico.
7-¿Cuál de la siguientes NO es indicación de intervención quirúrgica en un neumotórax?
a) Neumotórax bilateral simultáneo
b) Segundo episodio de neumotórax del mismo lado
c) Neumotórax en paciente EPCO con FEV1<1000cc.>20%
Síntomas
Recidiva
Hemoneumotórax
importante
Profesionales de
riesgo
NE bilateral
NE a tensión
Bullas en Rx
OXIGENO
REPOSO+OBSERVACIÓN OXIGENO
Drenaje pleural aspirativo
Reexpansión No reexpansión con
drenaje correcto.
No reexpansión
No fuga Fuga aérea
5º dia . Pinzar drenaje >7-8 días
y Rx control
Reexpansión
Retirada de drenaje
REEXPANSIÓN
ALTA
No reexpansión CIRUGIA
AUTORES:
Maria del Mar Vazquez Jiménez
Carlos Rueda Rios. Especialista en Neumología
Correspondencia con:
Carlos Rueda Rios
Hospital Universitario Virgen de la Victoria
MALAGA
Campus de Teatinos s/n
INDICE
I. Definición.
II. Clasificación.
III. Neumotórax espontáneo primario
1. Epidemiología
2. Clínica
3. Exploración física
4. Pruebas complementarias
5. Complicaciones
6. Tratamiento.
IV. Apéndice 1. Colocación del drenaje pleural.
V. Apéndice 2. Técnica de inserción del drenaje pleural.
VI. Apéndice 3. Manejo y criterios de retirada del drenaje pleural.
VII. Apéndice 4. Complicaciones del drenaje pleural.
VIII. Figuras.
IX. Bibliografía
X. Algoritmo
I. DEFINICION
El neumotórax se define como la presencia de aire dentro de la cavidad pleural que provoca el
colapso pulmonar del tejido adyacente.
II. CLASIFICACIÓN
Los podemos clasificar en función de la causa que los ha producido y en función de la
enfermedad pulmonar de base. Pueden ser espontáneos, traumáticos o yatrogénicos en
función de la causa y a su vez los espontáneos los podemos dividir en primarios o secundarios
en función de la patología respiratoria (tabla 1).
II.1 El neumotórax traumático se divide en cerrado o abierto en función de la
presencia o no de herida penetrante, con mucha frecuencia se acompaña de hemotórax de
cuantía variable. El barotrauma suele darse en pacientes sometidos a ventilación mecánica,
relacionándose este hecho con el uso de volúmenes corrientes y PEEP elevados.
II.2 Si no nos encontramos ante una causa clara estamos ante un neumotórax
espontáneo.
Yatrogénico:
• Tras cateterización de vías centrales, biopsia pleural,
toracocentesis, PAAF
Traumático:
• Heridas abiertas o cerradas
• Barotrauma
Espontáneo:
• Primario o idiopático
• Secundario (lesión pulmonar previo)
A/ Hablamos de neumotórax espontáneo primario si no encontramos ninguna
causa pulmonar de base que lo justifique y lo desarrollaremos más adelante.
B/ neumotórax espontáneo secundario, encontramos diversas situaciones
patológicas con afectación pulmonar, como son:
a) Asma: suele coincidir con las agudizaciones y con frecuencia se acompaña
de neumomediastino.
b) EPOC: con frecuencia son secundarios a la rotura de bullas intrapulmonares.
Suele cursar con importante afectación general y es conveniente sospecharla en
todo EPOC con aumento brusco de su disnea crónica y dolor pleurítico.
c) Fibrosis pulmonares avanzadas: en aquellas que existe panalizacion y bullas
como en el granuloma eosinófilo.
d) Enfermedades del tejido conectivo como síndrome de Marfan y Ehlers-
Danlos.
e) Neumotórax catamenial es una entidad rara que se da en mujeres de mediana
edad en el contexto de la menstruación y su sustrato anatomopatológico
corresponde a un foco endometriósico ectópico.
f) En cuanto a las causas infecciosas hay que destacar las neumonías
necrotizantes en especial la originada por el estafilococo, la tuberculosis que
puede originar focos caseosos subpleurales, el pneumocystis carinii , ...
g) Otras asociaciones son lo la fibrosis quiísticas, neumonías aspirativas,
infecciones fúngicas, sarcoidosis, carcinoma broncogenico, metástasis, etc.
III. NEUMOTORAX ESPONTANEO PRIMARIO
La causa más frecuente de rotura de pequeños blebs, que son colecciones de aire
subpleural resultado de ruptura alveolar, el aire diseca los tejidos conectivos adyacentes, acumulándose entre la lámina interna y externa de la pleura visceral. Los blebs se hallan con
más frecuencia en el vértice del lóbulo superior o en el vértice posterior del lóbulo inferior.
El neumotórax espontáneo es independiente del esfuerzo o la tos y se suele ver en varones
entre 20 y 40 años con tipo asténico y con hábito tabáquico. Se estima una incidencia de 7,4-
28 casos/100.000 habitantes en hombres y de 1.2-10 casos/100.000 habitantes en mujeres.
Es más frecuente en el lado derecho, los neumotórax bilaterales se producen en menos del
10% de los pacientes, en los dos primeros años recurren un 25% de los pacientes. Después del
segundo neumotórax, la posibilidad de tener un tercer episodio aumenta a más del 50%.
Las complicaciones más frecuentes en el desarrollo del neumotórax son el derrame pleural no
complicado en el 20%, el hemotórax con cuantía significativa en menos del 5% , el
neumotórax a tensión en un 2-3%, a mucha distancia el enfisema subcutáneo, el
neumomediastino , el empiema o la cronificación.
Las complicaciones postratamiento más frecuentes son la recurrencia y la fuga aérea
persistente que son indicaciones de cirugía.
III.1. CLINICA
Los síntomas depende de dos factores la reserva respiratoria del paciente y el tamaño
de neumotórax. Debido a que muchos de ellos son de pequeño tamaño y suceden en pacientes
sin patología pulmonar previa, pueden no provocar disnea debido a la reserva funcional del
paciente. Se estima que en un 5-10% de los casos pueden ser asintomáticos.
El síntoma más frecuente es la región torácica lateral, dolor en punta de costado, se
puede irradiar al resto del tórax y al cuello. Suele ser intenso, de carácter punzante y de inicio
agudo, con duración variable. Suele aumentar con la tos y movimientos respiratorios
profundos y la tos, aliviándose con la respiración superficial e inmovilización.
En algunos casos pueden existir manifestaciones vegetativas (sudoración, taquicardia,
palidez) u otro tipo de síntomas con tos seca, hemoptisis, síncope y debilidad de miembros
superiores.
III.2. EXPLORACIÓN FISICA
Cuando el tamaño del neumotórax es significativo encontramos disminución de los
movimientos de la pared del lado afecto, timpanismo a la percusión y disminución e incluso
ausencia de los ruidos respiratorios a la auscultación. En ocasiones podemos auscultar roce
pleural y taquicardia.
III.3. PRUEBAS COMPLEMENTARIAS
El diagnóstico clínico se confirma por radiografía posteroanterior y lateral de tórax al
identificar la línea del margen de la pleura visceral, separada de la pleura parietal existiendo
entre ambas un espacio aéreo hiperclaro sin trama vascular, de manera que el pulmón
adyacente se observa más denso a permanecer parcialmente colapsado (figura 5). En
neumotórax pequeños se puede realizar una posteroanterior de tórax en espiración forzada,
que identificará más fácilmente la línea de la pleura visceral. Es frecuente observar un
pequeño nivel hidroaéreo en el seno costofrénico, que no tiene ninguna relevancia clínica.
En ocasiones podemos observar que parte del parénquima permanece unido a pleura
parietal gracias a adherencias pleurales (figura 2 y 3)
En el electrocardiograma podemos observar una serie de signos característicamente
reversibles. En pacientes con neumotórax izquierdos el EKG puede mostrar una desviación a
la derecha del eje, con disminución de la amplitud del QRS e inversión de la onda T en
derivaciones izquierdas. En grandes neumotórax con la interposición de gas entre el corazón y
el electrodo puede producir cambios electrocardiográficos que simulen infartos anteriores.
III.4. COMPLICACIONES
A/ Neumotórax a tensión:
Se produce a raíz de que el aire pase del pulmón al espacio pleural durante la
inspiración y no salga por un mecanismo valvular. A medida que la presión en
el hemitórax aumenta, el mediastino se desplaza al lado contralateral interfiriendo con la ventilación, dificultando el retorno venoso y en último caso
disminuyendo el gasto cardiaco.
Debemos sospecharlo, cuando existe disnea intensa progresiva, taquipnea,
cianosis, taquicardia, hipotensión, diaforesis y distensión de las venosa
cervical.
Radiológicamente se observa desplazamiento contralateral de la tráquea y del
mediastino y depresión del diafragma ipsilateral
Si se sospecha el diagnóstico es imperativo el drenaje con aguja, catéter o tubo
sin confirmación radiográfica, antes de que se produzca el colapso circulatorio
completo. Una vez que ha sido solucionada la urgencia por la tensión, el
tratamiento ulterior debe ser similar al resto de pacientes con episodio
complicado.
B/. Hemoneumotórax:
Es una complicación poco frecuente, que se produce por rotura de
adherencias entre pleura parietal y visceral vascularizadas al producirse el
colapso pulmonar. A menudo la reexpansión pulmonar con drenaje ayuda a
taponar el lugar de sangrado. Si el sangrado no se controla o no se reexpande el
pulmon por la presencia de coágulos hay que valorar cirugía.
C/. Neumomediastino:
Se debe a que el aire pasa al mediastino que diseca a lo largo de los
bronquios y vainas vasculares de los vasos pulmonares, generalmente no tiene
consecuencias clínicas. No obstante debe descartarse otras causas como rotura
de la vía aérea o perforación de esófago.
D/.Enfisema subcutáneo:
No suele tener una implicación clínica significativa, si se produce tras el
drenaje indica que éste está mal posicionado u obstruido, o bien uno de los
orificios de drenaje se halla en pared torácica.
E/.Neumotórax bilateral:
El neumotórax bilateral simultáneo es raro (menor del 1%) y precisa, en
la fase aguda, drenaje de ambos espacios pleurales. Es más frecuente el
neumotórax bilateral secuencial (recidiva contralateral) que es indicación.
F/.Pioneumotórax
Generalmente es secundario a neumonía necrotizante o a una rotura
esofágica. Es indicación de drenaje urgente.
G/.Neumotórax crónico
Se trata de un neumotórax que persiste más de tres meses. Suelen
perdurar gracias a la existencia de adherencias pleurales, fístula broncopleural
a través de una bulla o alteración parenquimatosa (necrosis, nódulo, etc). La
cavidad se suele rellenar de líquido. Suele tratarse con intervención quirúrgica.
III.5. TRATAMIENTO
Va a depender de varios factores: tamaño del neumotórax, enfermedad pulmonar
previa, causa, síntomas recidiva, tratamientos previos, profesiones de riesgo, etc., puede ir
desde la observación hasta el abordaje quirúrgico. Las recidivas ocurren en un 30 a 50% de
los NEP, y en un 80% de los casos se producen durante el primer año.
El tratamiento debe cumplir dos objetivos:
1-evacuar el aire de la cavidad pleural
2-toda vez que el aire halla sido drenado, conseguir una reexpansión duradera y
estable que evite las recidivas.
Hay una serie de medidas generales que hay que imponer a todo neumotórax sea cual
sea su tamaño y es el reposo y la oxigenoterapia, que ayudan a acelerar la reabsorción del aire
intrapleural.
En neumotórax espontáneo primario podemos ofrecer cinco tipos distintos de tratamiento:
• Reposo y observación clínica. en régimen de ingreso hospitalario o en domicilio
paciente colaborador, proximidad geográfica, cuando se trata de un primer episodio de
neumotórax espontáneo primario, menor del 20% y asintomático.
• Drenaje pleural: indicado en el primer episodio de los NEP mayores del 20% y en
todos los neumotórax espontáneos secundarios.
• Pleurodesis
• Cirugía mediante toracotomía con pleurectomía parietal parcial o abrasión mecánica
• Cirugía videotoracoscópica.
1. Reposo y observación clínica.
Cuando se trata de un primer episodio de neumotórax espontáneo primario, menor del
20% y asintomático, en pacientes sin enfermedad respiratorio de base pueden ser tratados de
forma expectante con reposos y preferiblemente observación hospitalaria durante las primeras
24-48 horas. Posteriormente se hará un seguimiento clínico-radiológico ambulatorio.
Hay grupos que están planteando en tratamiento expectante en domicilio. Se lleva a cabo
en paciente colaborador y con criterios de proximidad geográfica, deberá permanecer en
reposo con oxigenoterapia continua con posibilidad de seguimiento en consulta.
El principal inconveniente de esta opción terapéutica es la progresión del neumotórax o la
no reexpansión, en cualquier caso si pasado una semana el neumotórax ha progresado o no se
ha reexpandido el pulmón se recomienda colocación de drenaje.
2. Drenaje pleural:
Indicado en el primer episodio de los NEP mayores del 20% , en los a tensión en herida
abierta del tórax, bilateral, sintomático, enfisema subcutáneo, neumomediastino, progresión radiográfica, caso de ventilación mecánica y en todos los neumotórax espontáneos
secundarios.
La técnica de inserción, seguimiento y criterios de retirada se explicaran más adelante
Los tubos de drenajes finos, pueden utilizarse en neumotórax espontáneo primario, sobre
todo si es el primer episodio y si el neumotórax no se acompaña de traumatismo, hemotórax,
hidrotórax ni disfunción respiratoria. Los tubos de drenajes se conectan a sistemas
unidireccionales que no permitan la entrada de aire en cavidad pleural, el más utilizado es el
de tres cámaras.
3. Pleurodesis:
Se reserva para pacientes que no puedan ser sometidos a toracotomía (mala calidad de
vida previa, edad avanzada, neoplasias con progresión pleural,...). Su objetivo es conseguir a
unión de ambas pleuras con distintos agentes (talco, clorhidrato de tetracicilina, bleomicina o
colas biologicas) que se administran mediante drenaje pleural o toracoscopia.
4. Cirugía:
Se realiza por minitoracotomía axilar con pleurectomía parietal parcial o abrasión pleural,
que a diferencia de la anterior preserva el plano extrapleural. En los últimos años gracias al
desarrollo de las suturas mecánicas la resección de bullas es mucho más efectivo.
5. Cirugía videotoracoscópica (CVT):
Con resultados similares a la cirugía convencional, pero es menos agresiva y tiene un
periodo postquirúrgico más corto. Consiste en la realización de por lo menos tres orificios en
el hemitórax afecto por los que se introduce una cámara y el instrumental.
Las ventajas de la cirugía videotoracoscopica se puede cifrar en cuatro:
1. Permite una mejor visualización de toda la superficie pleural
2. Es más rápida de realizar.
3. Produce menos dolor postoperatorio
4. Disminuye la estancia hospitalaria.
APENDICE 1. COLOCACIÓN DE DRENAJE PLEURAL
SECUENCIA MATERIAL
PREPARACIÓN
Explicación
Posición sentado o decúbito supino
Atropina 0,5 mg IM
Localización del sitio de punción
Marcar el lugar de punción
CAMPO ESTERIL
Povidona yodada, paño, guantes y gasas
esteriles
ANESTESIA LOCAL
Agujas IM, Jeringas de 10 ml,
Escandicaina
MATERIAL DE DRENAJE
Tubo de drenaje: convencionales de silastic con múltiples orificios, con línea radiopaca hasta el último agujero de varios diámetros, tipo Pleurecath catéter fino y largo con llave de tres pasos y el Neumovent catéter fino y corto que lleva incorporada una válvula unidireccional, pinzas de Kelly, sutura, porta, sistema de sellado de agua y aspirador.
APENDICE 2. Técnica de inserción del tubo de drenaje.
Se coloca al paciente sentado o en decúbito supino se elige la línea medioclavicular en
2ª espacio intercostal (aire) o en línea axilar media 5ª-6ª espacio intercostal (liquido o aire).Se
realiza la limpieza y antisepsia de la piel.
Los tubos finos tienen un fiador interno o externo y sólo tienen que ser empujados a
través de la zona previamente anestesiada hasta llegar a cámara pleural. Se dirige la punta del
catéter al apice y se extrae el fiador. Se fija en tubo con un punto y se conecta al sistema de
vacío a través de la llave de tres pasos.
En el tubo de drenaje convencional se anestesia con rutina habitual y se realiza una
incisión en piel y se diseca por planos con pinza de Kelly debiendo ir siempre por borde
superior de la costilla. Se introduce suavemente el tubo de drenaje, se debe comprobar
presiones intrapleurales, fijar drenaje, se conecta a sistema de sellado de agua (pleurevac) y se
comprueba colocación mediante radiografía.
Para el líquido se prefiere una localización en 5º o 6º espacio intercostal en línea axilar
media, deben evitarse localizaciones más bajas por el riesgo de atravesar diafragma y lesionar
órganos intraabdominales, evitaremos si es posible la colocación en la espalda, ya que
técnicamente es más complicado y produce un innecesario dolor al enfermo.
Posteriormente se conecta a aspirador caso de tratarse de un neumotórax o si no evacua de
forma efectiva en derrame pleural (nunca mas de 1000 ml/8h)
APENDICE 3. Manejo y criterios de retirada de drenaje pleural
Manejo
Tras la inserción del tubo se comprueba posición y la marcha de la reexpansión pulmonar con
una radiografía. Debe ser conectado a un sistema que impida la salida de aire pero no su
entrada, estos sistemas se dividen en dos bloques: pasivo (válvula de Heimlich) o activo
(recolector de tres cámaras) El tubo nunca debe ser pinzado para el traslado de los enfermos,
especialmente si tiene fuga aérea por el peligro de que se convierta en neumotórax
hipertensivo. .
Si un drenaje torácico se desconecta accidentalmente simplemente debe ser reconectado.
Las pequeñas oscilaciones de los líquidos del tubo indican que están permeables si no se
mueven o el pulmón se halla reexpandido o el tubo obstruido. Grandes oscilaciones se asocian
a atelectasia o reexpansión incompleta.
Para repermeabilizar un tubo puede ordeñarse, pero no se recomienda la irrigación con suero
salino por el peligro de contaminación.
Criterios de retirada
Utilizamos tres criterios clásicos fundamentales :
• no hay oscilaciones de líquido
• el drenaje es menor de 100 cc por día
• no hay fuga aérea.
Se pinza el tubo durante 24 horas y si no se ha reproducido el neumotórax se extrae el
tubo. Para cerrar el orificio debe anudarse el punto que se dejó con este propósito (tubos de
calibre convencional) o gasas con vaselina en los tubos de menor calibre. Tras la retirada debe
hacerse una radiografía de control.
Sistema de tres cámaras:
La primera recoge el líquido, la segunda actúa como sello de agua que no permite la
entrada de aire y permite su salida y la tercera es la cámara de control de succión, que en los
sistemas secos se ha sustituido la columna de agua por un sistema valvular.
Este sistema se conecta a su vez a la red de presión negativa del hospital o a los sistemas
portátiles de aspiración.
APENDICE 4. Complicaciones de los drenajes
a) Fallo de la reexpansión y cronicidad: Nos ocurrirá caso de reexpansión incompleta a pesar
de la buena colocación y aspiración del tubo. En este caso se requiere toracotomía para
valorar la decorticación.
b) Fuga aérea persistente: Ocurre en aproximadamente un 4% de los drenajes. Generalmente
si esta situación se prolonga más de 7-10 días habría que valorar cirugía, caso de que se
acompañe de reeexpansión incompleta hay que valorar un segundo drenaje.
c) Colocación incorrecta: Lo mas frecuente es en los tubo de pequeño calibre al dejar algún
orificio de drenaje fuera de cavidad pleural, provocando enfisema subcutáneo, más
infrecuente es colocarlo en los tejidos blandos de cavidad torácica. En cualquiera de estas dos
situaciones habría que recolocar el tubo.
c.1 Caso de perforación pulmonar, diafragma u órgano intraabdominal habrá que
valorar cirugía urgente.
c.2 Caso de colocar un tubo dentro de una bulla habrá que operar al paciente
inmediatamente.
d) Enfisema quirúrgico: casi siempre ocurre por una colocación incorrecta del tubo o bien por
obstrucción del sistema de aspiración.
e) Hemorragia intrapleural: Suele ocurrir más en ancianos y se da por colocar en tubo justo
bajo la costilla.
f) Empiema: Se produce más en neumotórax secundarios a traumatismos o casos de drenajes
mantenidos durante tiempos prolongados.
g) Edemas de reexpansión: Es una complicación rara pero potencialmente letal. Se produce
cuando el aire o el líquido se evacuan demasiado rápido por un aumento de la permeabilidad
capilar. Los síntomas son tos progresiva, dolor pleurítico y edema bilateral. Para prevenirlo
debemos pinzar el tubo periódicamente para evitar la reexpansión súbita.
h) Neuralgia intercostal: Presenta dolor irradiado anteriormente, y en ocasiones se cronifica
mucho después de la retirada del drenaje.
FIGURA 1
BULLA VISUALIZADA POR TORACOSCOPIA.
16
FIGURA 2
Figura 2. ADHERENCIAS PLEURALES
17
FIGURA 3
NEUMOTORAX SECUNDARIO CON
ADHERENCIA PLEURAL.
18
FIGURA 4
NEUMOTÓRAX DEL 100% PULMON IZQUIERDO CON
DESPLAZAMIENTO MEDIASTINICO
CONTRALATERAL.
19
FIGURA 5
NEUMOTORAX ESPONTÁNEO PRIMARIO.
20
fIGFIUGRURA 6
DISPOSITIVO DE DRENAJE DE 3 CAMARAS.
21
FIGURA 7
Esquema de funcionamiento de la válvula unidireccional de Heimlich.
22
BIBLIOGRAFIA
• Baumann MH, Strange C. The Clinician's Perspective On Pneumothorax Management.
Chest. 1997; 112(3). 822-8.
• Ginferrer Garolera JM, Fernander-Retana P, Rami Porta R. Tratamiento del neumotorax
espontáneo mediante drenajes de pequeño calibre. Arch Bronconeumol. 1990; 26 144-
146
• Sanchez-Lloret J. Canto A, Borro Jm, Gimferrer JM. Diagnóstico y tratamiento del
neumotórax. Arch Bronconeumol 1998; 34 Supl 3:24-30
• Schramel FM, Postmus PE, Vandrschueren RG. Current aspects os spontaneous
pneumothorax. Eur Respir J 1997; 10: 1372-1379
• Sahn SA, Heffner Spontaneuous pneuomothorax. N Engl J Med 2000; 342:868-873
• Rueda Ríos C, Tibos F Hidalgo Sanjuán MV. Hemoptisis catamenial. A propósito de un
caso. Arch Bronconeumol. 2000; 36: 539
23
CUESTIONARIO
1. En un paciente con clínica de dolor de carácter pleurítico y disnea de aparición súbita, sin
antecedentes respiratorios de interés en un centro de salud que aporta radiografía de tórax
AP en inspiración normal. En la exploración la auscultación es normal. Cual sería la
actitud más correcta:
a) Oxigenoterapia y broncodilatadores.
b) Colocar un drenaje pleural tipo abocath en hemitórax afecto.
c) Tratar con benzodiacepinas
d) Realizar radiografía de tórax en espiración forzada
e) Realizar electrocardiograma.
2. Un enfermo con un cáncer de colon que precisa la canalización de una vena subclavia para
la su ciclo quimioterápico, comienza a presentar disnea y dolor torácico de carácter
pleurítico. En la exploración el enfermo presenta cianosis, frecuencia respiratoria de 35
rpm y disminución del murmullo vesicular en hemitórax derecho ¿diagnostico más
probable?
a) Reacción alérgica a la quimioterapia.
b) Tromboembolismo pulmonar
c) Angina
d) Neumotórax
e) Trombosis de la vena subclavia.
3-Paciente de 16 años que presenta un neumotórax izquierdo de menos de un 15% sin
repercusión respiratoria. Hace 2 años presentó un neumotórax derecho. Señale cual sería el
tratamiento más adecuado:
a) Pleurodesis química con talco
b) Drenaje torácico
c) Observación y cirugía posteriormente
d) Profilaxis antibiótica para evitar la infección de la cavidad pleural.
f) Pleurodesis con urokinasa.
24
4-Paciente de 20 años neumotórax con colapso pulmonar del 40%, con antecedente de un
episodio previo del mismo lado:
a) Drenaje torácico
b) Drenaje torácico y posterior intervención quirúrgica
c) Pleurodesis química con talco
d) Observación durante 24 horas y si el colapso pulmonar no aumenta al alta domiciliaria
e) Oxigenoterapia de alto flujo.
5-A los 20 minutos de una intervención quirúrgica en la que se requirió ventilación mecánica
y aporte de fluidos por vía central , un enfermo presenta tos seca, disnea y sensación de
opresión torácica súbitamente. En la exploración se aprecia disminución del murmullo
vesicular en hemitorax derecho. Diagnóstico.
a) Atelectasia masiva
b) TEP
c) Neumotórax
d) IAM
e) Crisis de ansiedad postquirúrgica
6- En relación al neumotórax, es FALSO que
a) Los neumotórax tienen una alta frecuencia de recidivas.
b) Los neumotórax espontáneos primarios suelen ocasionarse por rotura de blebs
apicales.
c) La causa más frecuente de neumotórax espontáneo secundario es EPOC.
d) Ante la sospecha de neumotórax debe solicitarse una radiografía en inspiración
forzada
e) Los neumotórax espontáneos primarios no tienen relación con el hábito tabáquico.
7-¿Cuál de la siguientes NO es indicación de intervención quirúrgica en un neumotórax?
a) Neumotórax bilateral simultáneo
b) Segundo episodio de neumotórax del mismo lado
c) Neumotórax en paciente EPCO con FEV1<1000cc.>20%
Síntomas
Recidiva
Hemoneumotórax
importante
Profesionales de
riesgo
NE bilateral
NE a tensión
Bullas en Rx
OXIGENO
REPOSO+OBSERVACIÓN OXIGENO
Drenaje pleural aspirativo
Reexpansión No reexpansión con
drenaje correcto.
No reexpansión
No fuga Fuga aérea
5º dia . Pinzar drenaje >7-8 días
y Rx control
Reexpansión
Retirada de drenaje
REEXPANSIÓN
ALTA
No reexpansión CIRUGIA






